|
The information view is an HTML-based interface, which displays all information generated based on the loaded nsSNP data. Currently, BALL-SNP generates for each nsSNP input file:
Database information
Experimentally gained knowledge about nsSNPs is deposited and curated in different databases. Some of these databases provide additional information concerning the pathogenicity and clinical significance of a nsSNP. To make use of this knowledge, we include information from
 Besides, we also include information concerning available drug targets deposited in the DrugBank. Active site calculation
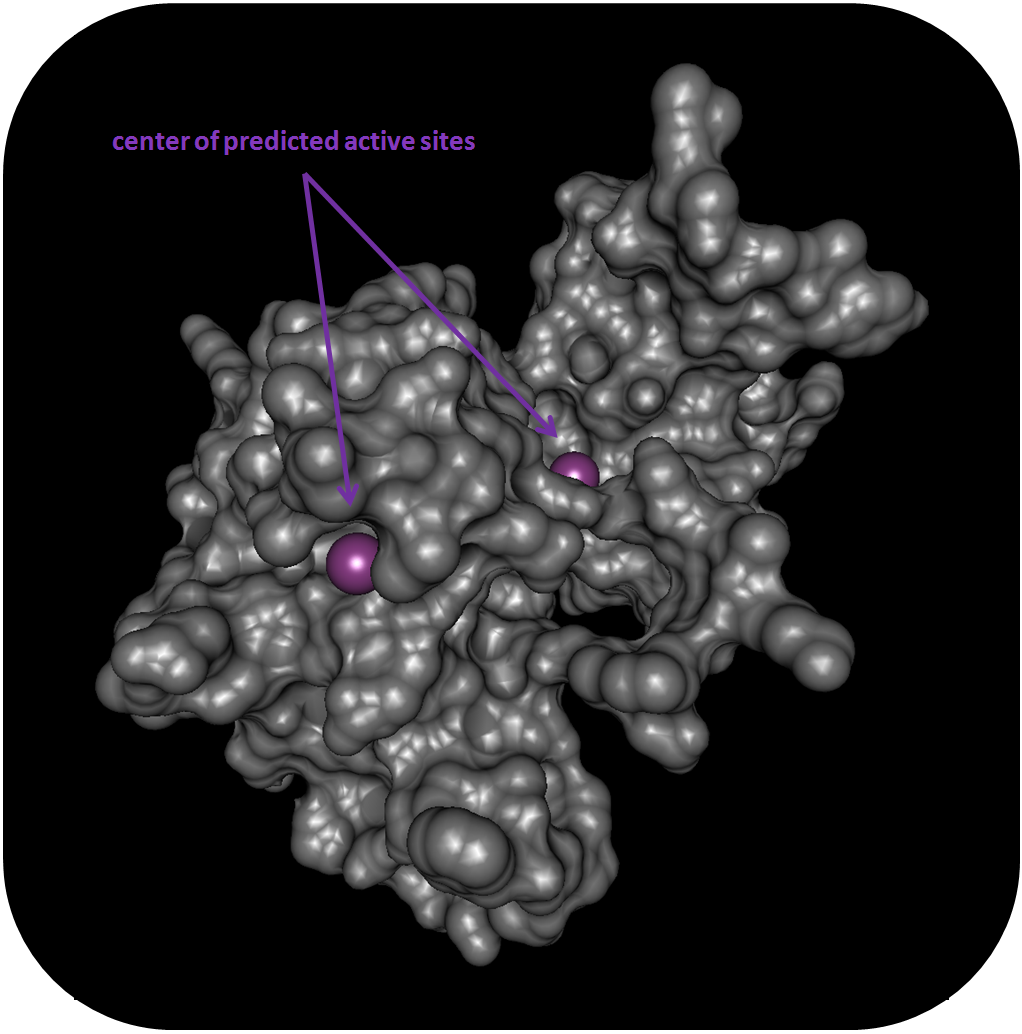
In addition to known information on pathogenicity from databases,the proximity of nsSNPs to functional sites such as binding pockets for ligands plays a crucial role.BALL-SNP predicts active sites, which often are located in the largest surface cleft, based on the Putative Active Sites with Spheres (PASS) method, that use probe spheres to characterize regions of buried volume on a protein surface. Based on size, shape, and burial extent of these volumes, positions, which putatively represent binding sites, are identified. The predicted active sites are visualized as spheres in BALL-SNP, which represent their centers. These spheres are visualized as large atoms in purple color.
 Cluster analysis
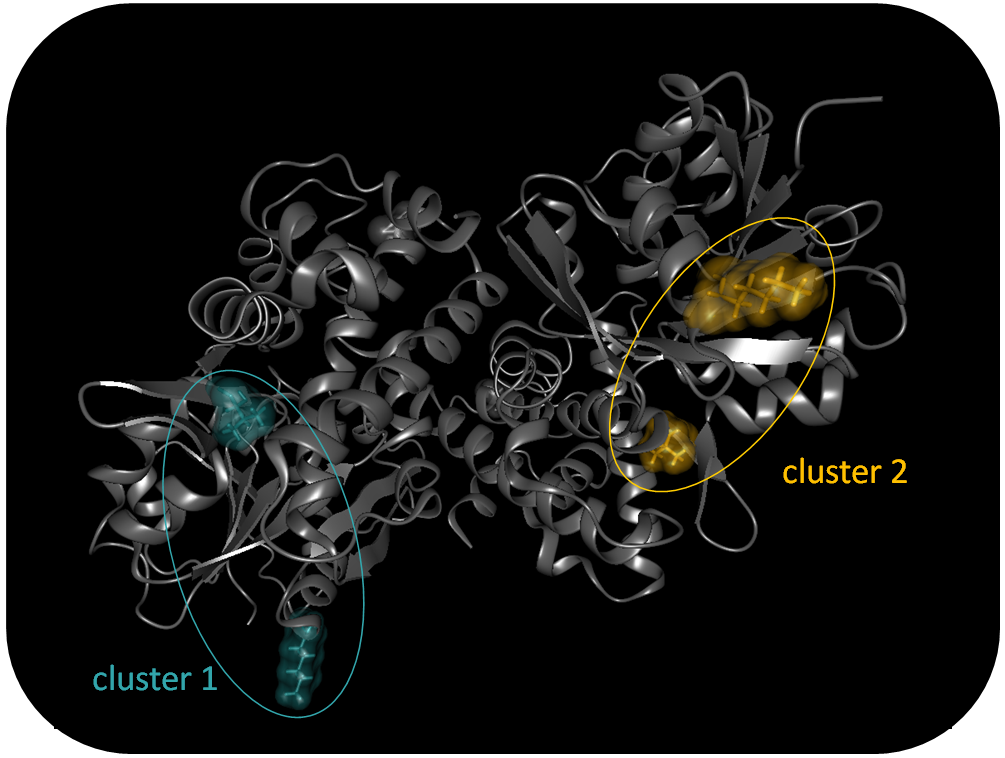
To identify nsSNPs with putative synergetic effects, we perform a hierarchical bottom-up cluster analysis based on the C-alpha atom distances of all mutated residues. Users can select a threshold for cluster affiliation and the comprised residues are colored in the 3D View, accordingly, via links in the information view.
 Nearest neighboring nsSNP
To identify clustering nsSNPs not only visually in the 3D View, we calculate the pairwise C-alpha atom distances of all mutated residue. The nsSNPs with shortest C-alpha distance are also listed in the information view table to support the visual inspection.
Protein stability prediction
Proteins properly folded have minimal potential energy and are usually stable. Amino acid substitutions introducing a change in the protein sequence can have a significant impact on the potential energy of the protein structure, and thus its folding and stability. Consequently, the analysis to which extent a mutation affects protein stability with respect to the wild type, extends the understanding of the mutation impact on protein function and the genotype-phenotype relationship, accordingly.
We currently use freely available I-Mutant 2.0 code. I-Mutant 2.0 automatically predicts protein stability changes caused by single point mutations in protein sequence using support vector machines (SVMs). Pathogenicity prediction
Several in silico tools to predict the functional impact of nsSNPs on a protein’s function are available. To make use of their functionality, we include currently:
The prediction methods have been selected depending on stand-alone availability and different prediction strategies. Pathogenicity consensus
Based on the parsed information from ClinVar and HUMSAVAR, the predicted protein stability changes as well as the pathogenicity prediction results, BALL-SNP calculates a simple majority vote-based consensus for all comprised nsSNP-introduced amino acid substitutions. The consensus can assume the following values:
 Users can color the mutated residues within the 3D View according to the calculated pathogenicity consensus via a link in the information view. 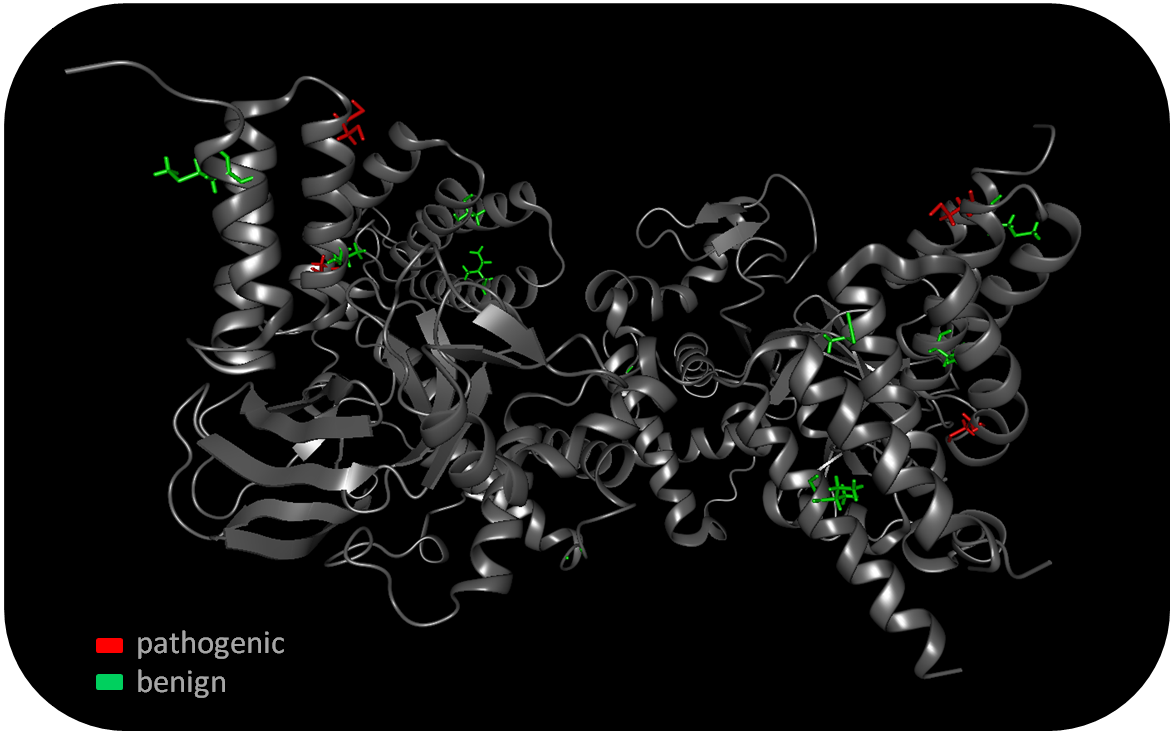 If a disease-associated information is available in one of the included databases, this information is prioritized and the pathogenicity consensus score is set to pathogenic for the corresponding nsSNP-introduced amino acid substitution.
Back to BALL-SNP tutorial main page.
|