Genetic alterations such as non-synonymous single nucleotide variants (nsSNVs), single base changes in DNA coding regions altering a protein’s amino acid sequence, play a critical role in human diseases.
In Mendelian disorders, where variation in a single gene is responsible for the phenotypic consequence, thousand causative genes have been identified already. Detecting the causative genes in common diseases such as hypertension or diabetes, however, still remains a challenge. In general, these diseases are caused by a varying number of genetic alterations and are influenced by environmental factors that modulate the severity and type of disease-related phenotypes.
Since the experimental analysis of the pathogenic potential of variants is laborious and time-consuming, several methods to predict the biological impact of nsSNVs on the corresponding proteins and their function have been developed in the last decade. However, none of these methods actually considers the influence of several nsSNVs or mutual effects. From a medical point of view, especially the individual combination of a human individual’s nsSNVs may play a crucial role in clinical diagnostics regarding personalized medicine, since genetic variations for example have been identified to influence selection, dosing and adverse events of medical drugs.
Traditional approaches predict the influence of nsSNVs on pathogenicity for single variants. However, the impact of nsSNVs on a patient’s phenotype can arise from multiple factors. Accumulating nsSNVs may have a quantitative effect on a gene’s dysfunction as well as nsSNVs in interacting genes may aggravate or attenuate a pathological effect. In consequence, a multi-scale analysis of nsSNVs, which includes three-dimensional context, interaction information and functional cascades, is required to capture the whole range of nsSNV impact.
To address this issue, we determined putative nsSNVs associations in a high-quality clinical data set of cardiomyopathy patients and studied possible accumulation events affecting a patient’s phenotype. We further analyzed available protein three-dimensional structures within our data set and the impact of nsSNVs to gain insights in pathogenic molecular mechanisms.
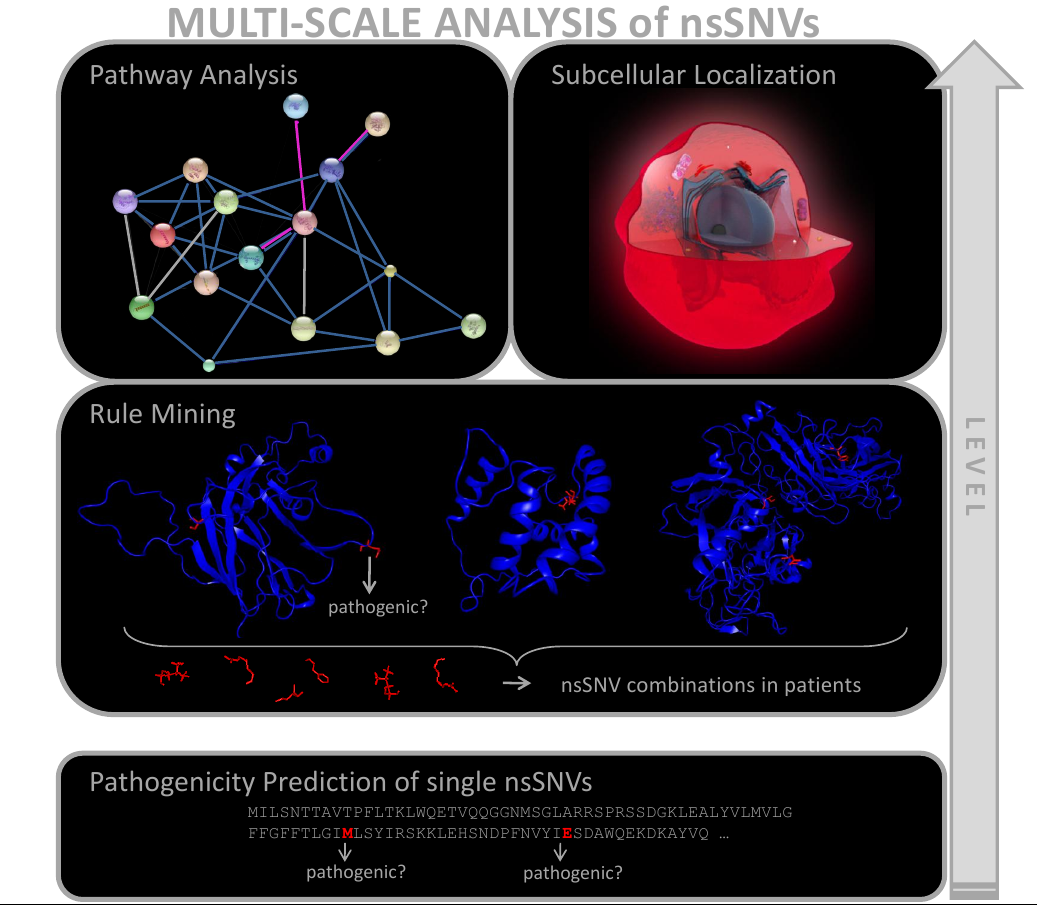
A detailed description of the developed pipeline and the conducted analysis of cardiomyopathy patients is currently prepared for publishment. The cardiomyopathy data was previously published in the “Atlas of the clinical genetics of human dilated cardiomyopathy”. The corresponding sources and information of the pipeline can be downloaded below:
Source: multi_scale_src