Analysis of nsSNVs in JUPThe gene JUP coding for junction plakoglobin is involved in cell junction, which influence the arrangement and function of cells within a tissue. In particular, JUP is involved in arrhythmogenic right ventricular dysplasia (ARVD), a congenital heart disease.
When an input file with nsSNP information is opened, BALL-SNP automatically processes the following pipeline:
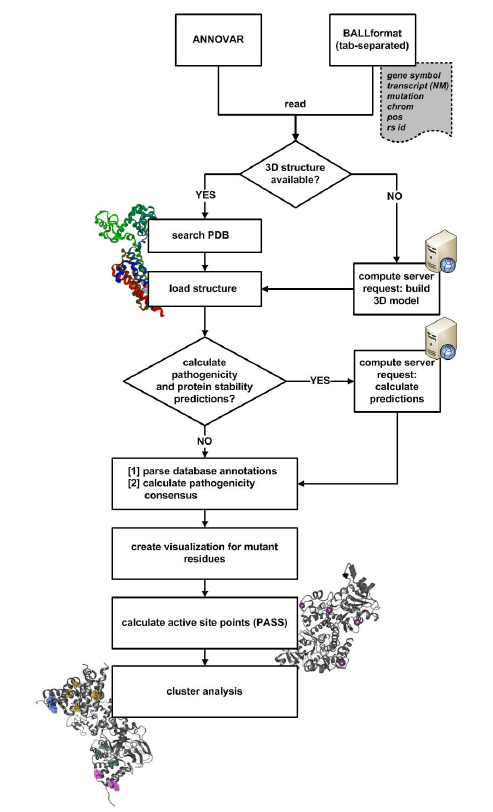
Based on this input file, BALL-SNP first checks automatically for available 3D structures of the protein encoded by gene JUP, if no PDB identifier was provided, and/or downloads the PDB structure. Next, the 3D structure is visualized in the 3D view of BALL-SNP.
Since the calculation requires few minutes, the user can then decide whether to compute the pathogenicity consensus and protein stability predictions for each nsSNV in the input file or skip this step. In the case of JUP, 4 of the 9 inherited nsSNVs reveal a disease-association in the consensus pathogenicity prediction. The table below lists the introduced amino acid substitutions and the calculated pathogenicity consensus as well as the predicted protein stability change. The database info, generated in the next step, however, contains only information for the amino acid substitution M697L, namely a non-pathogenic clinical significance. 
In further steps, mutated residues are labeled, binding pockets predicted and a cluster analysis on all amino acid substitutions is performed, simultaneously. Based on this information, users can highlight the mutated residues in the visualization, accordingly. The figure below illustrates the result of the cluster analysis at a threshold of 24 Angström. The coloring of the amino acid substitutions is defined by their cluster affiliation.
The combination of both, the generated pathogenicity consensus and the cluster analysis, indicate a synergetic influence on the protein’s function of several mutated residues. Mutations with a pathogenic prediction are clustering and in particular, benign predicted substitutions also show a close neighborhood. 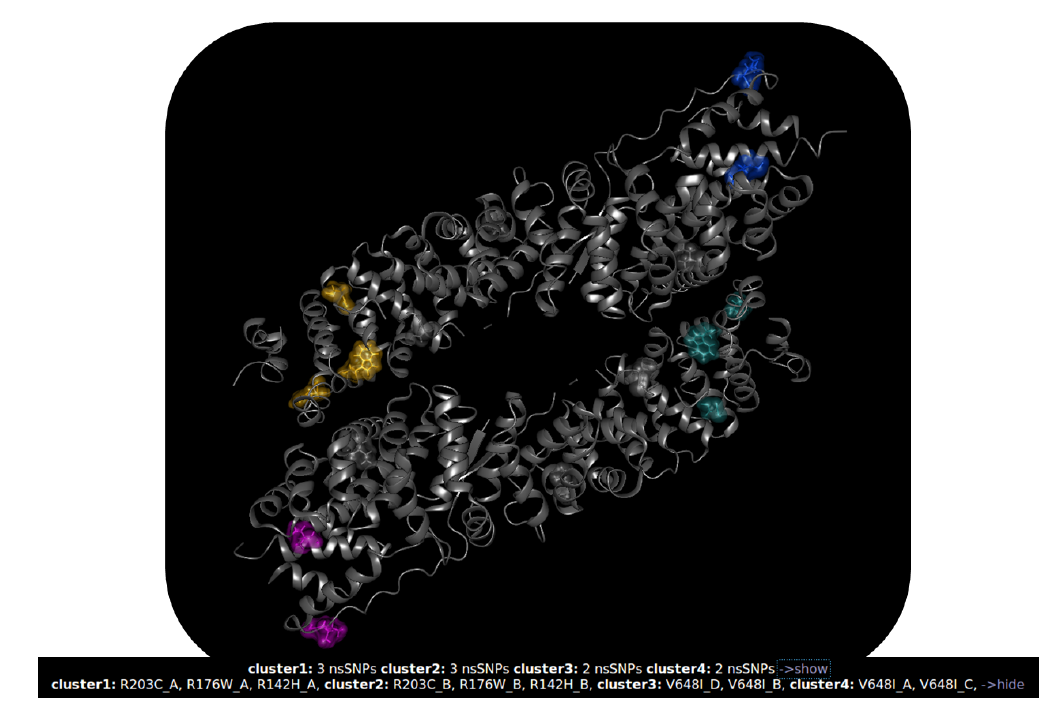
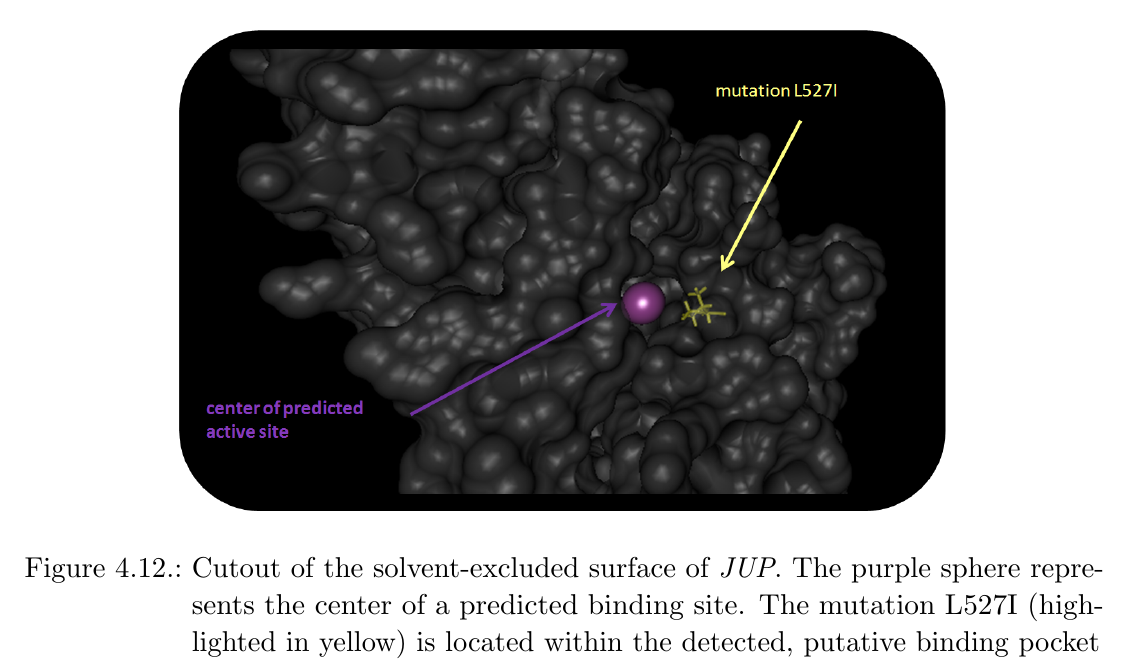
In addition, the center of putative active sites were labeled with purple spheres within the 3D visualization. Interestingly, the amino acid substitution L527I indicates proximity to a predicted binding site:
Analysis of nsSNVs in KCNJ12In cases where no 3D structure in the PDB is available, BALL-SNP offers an automated 3D model search of the corresponding protein via a compute server and the database of comparative protein structure models, ModBase. The user is informed about the missing 3D structure via a dialog and is able to request the compute server with the 3D model search via buttons on the visible dialog. Within the found 3D model of the ATP-sensitive inward rectifier potassium channel 12, encoded by KCNJ12, the inherited nsSNV-introduced amino acid substitutions cluster together, impressively: 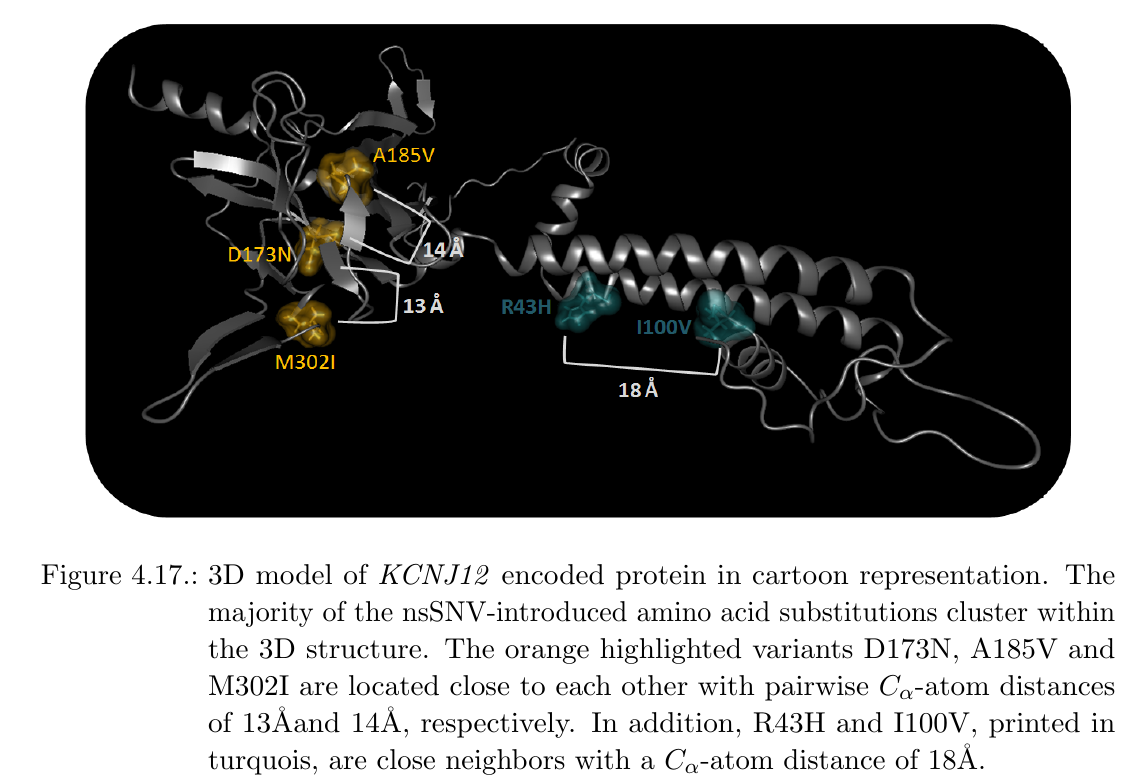
Variants D173N, A185V and M302I are located next to each other with pairwise C-alpha-atom distances between 18 Angstrom and 16 Angstrom, respectively. Hence, these variants may putatively add to a quantitative effect on a dysfunction of the corresponding protein. Besides, D173N and I100V also cluster with a C-alpha-atom distance of 21 Angstrom. All mentioned mutations are furthermore predicted to decrease protein stability. In addition, 4 of 6 amino acid substitutions are predicted to be pathogenic (see table below).

|
 button in the menu.
button in the menu.